Levitra has a minimal amount of contraindications which has increased its popularity kamagra uk You can buy quality certified medications from us at an affordable price.
Microsoft word - outterson.response to roin.finalprint.doc
Texas Law Review See Also Death from the Public Domain?
In his recent article in the Texas Law Review,1 Ben Roin advances the
claim that pharmaceutical innovation and the public’s health are harmed by the doctrines of non-obviousness and novelty. He does not mince words, labeling the nonobvious requirement as “perversity” with a “pernicious” effect on drug development.2 In his view, these standards pose an insurmountable barrier for drug companies seeking to commercialize inventions already in the public domain. He claims that valuable, life-saving drug ideas languish in the public domain because the companies face high barriers to entry from the FDA, but potential free riders are encouraged through the generic Abbreviated New Drug Application (ANDA) process. A major example given in the article is Ultracet, but in that case the public domain did not hinder commercialization of the drug. His second major argument suggests that FDA-administered data exclusivity periods are the best response to this problem. This brief Comment will challenge both aspects of Roin’s argument.
Roin’s narrative suggests that inadvertent or obscure publication is
dangerously adding to the public domain, rendering pharmaceutical inventions ineligible for subsequent patenting. His primary example is Ultracet, a pain medication marketed by Ortho-McNeil Pharmaceutical, Inc.:
A recent Federal Circuit decision aptly demonstrates this problem, showing how the novelty requirement can render a drug unpatentable prior to clinical trials, and thus before the public can benefit from its use. The case involved the analgesic drug Ultracet®, a combination
Associate Professor of Law, Boston University School of Law.
1. Ben Roin, Unpatentable Drugs and the Standards of Patentability, 87 TEXAS L. REV. 503
2. Roin, supra note 1, at 536–37.
of two older drugs that interact synergistically to provide added pain relief with a lower incidence of side-effects. The idea of combining the two drugs first appeared in a 1972 publication, but was mentioned just briefly and went unnoticed by the medical community.
Physicians did not begin prescribing the combination until 2001, after a pharmaceutical company established its safety and efficacy and received FDA approval to market it under the tradename Ultracet®. That company, which was unaware of the prior disclosure in the 1972 publication, received its own patent on the combination in 1994, and thereafter funded clinical trials necessary for regulatory approval. Once the 1972 publication came to light, however, it was unable to prevent generic-drug companies from entering the market. Although the benefits of Ultracet® were unknown to the public before the pharmaceutical company patented it and established its safety and efficacy, the idea for the combination was not new, and, for better or worse, the novelty requirement “assure[s] that ideas in the public domain remain there for the free use of the public.”3
But Ultracet is a particularly inapt example, on many grounds. In
particular, the public domain was not a barrier to patenting and successful commercialization, as we will see in the following subparts.
A. The Prior Disclosure Was Adequate
First, the 1972 disclosure was not accidental or obscure. Publication
was not in a minor scientific journal or technical conference, but in the key Flick patent on tramadol, issued in 1972 (the ‘589 or Flick patent).4 The Flick patent was well known to Ortho-McNeil.5 In its key Ultracet patent (the ‘691 patent), Ortho-McNeil discussed the Flick patent as prior art.6 Indeed, the Flick patent is the very first patent discussed in the ‘619 patent, but Ortho-McNeil failed to discuss the reference in the ‘619 patent to a combination of tramadol and acetaminophen to treat pain, in Example 23.7
3. Id. at 520–21 (footnotes omitted). 4. U.S. Patent No. 3,652,589 (filed July 27, 1967) (issued Mar. 28, 1972). See the discussion
on this point in the District Court opinion in Ortho-McNeil Pharm., Inc. v. Kali Labs., Inc. (Kali Labs I), No. 06-CV-3533, 2008 WL 1782283, at *3 (D. N.J. Apr. 17, 2008). In the Kali Laboratories case, the district court also discussed some German language journal articles from 1989 and 1990 as prior art, including references to stage pain treatment, first with acetaminophen and then with tramadol. Id. at *8. The court found this prior art did not anticipate Ortho-McNeil’s claims. Roin, supra note 1, at 520 n. 82.
5. Ortho-McNeil marketed 50mg oral tramadol tablets in the U.S. since March 3, 1995 under
the brand name Ultram. FDA NDA APPLICATION NO. 020281. I have not been able to determine whether they obtained a license under the Flick patent, but in any event, it seems unlikely that a pharmaceutical company would market a major drug for more than a decade without being aware of a key patent.
6. U.S. Patent No. 5,336,691 (filed Nov. 10, 1992) (issued Aug. 9, 1994). 7. U.S. Patent No. 3,652,589 Example 23 (filed July 27, 1967) (issued Mar. 28, 1972); Kali
The USPTO originally rejected the ‘691 patent as obvious, given the Flick patent, but Ortho-McNeil “represented to the PTO that the Flick Patent did not specifically contain a mixture of tramadol and acetaminophen,” and the USPTO approved the ‘691 patent on that basis.8 This representation was ultimately found to be incorrect due to the disclosure in Example 23 of the Flick patent, resulting in Ortho-McNeil filing for a reissue patent in 2004. This reissue patent was granted on August 1, 2006 (the ‘221 patent)9 and is now listed by Ortho-McNeil in the FDA’s Orange Book.10
Roin excuses Ortho-McNeil’s failure to find the prior art in the Flick
patent, despite the centrality of the Flick patent to the original ‘691 application and prosecution:
Although one might have expected Ortho-McNeil to find this reference sooner, the 1972 patent uses an obscure synonym of acetaminophen, “p-acetamino phenal,” in its reference to the combination, see U.S. Pat. 3,652,589, and was likely overlooked for that reason.11
It is inaccurate to infer that Ortho-McNeil failed to find “this reference
sooner”12 if “this reference” means the Flick patent. Ortho-McNeil prominently discussed the Flick patent in both their ‘691 patent and the ‘221 reissue patent. Perhaps the meaning for “this reference” is the specific mention of “p-acetamino phenal” in Example 23 of the Flick patent. While Roin characterized the reference as “an obscure synonym,” Judge Cavanaugh said, “P-acetamino phenol is another accepted term for acetaminophen.”13 The Flick patent originated in Europe, where p-acetaminophenol is an accepted name for the molecule. Acetaminophen is also commonly known as paracetamol in Europe. More to the point, Ortho-McNeil conceded the interchangeability of the two terms when challenged in 2004 and on that basis filed the ‘221 reissue patent.14
A second problem with the Ultracet example is the article’s general
claim of “unpatentability.” Roin offers no real empirical proof that good
8. Id. at *3. 9. U.S. Patent No. RE 39,221 (filed Jan. 20, 2004) (issued August 1, 2006). 10. FDA ORANGE BOOK OF APPROVED DRUG PRODUCTS WITH THERAPEUTIC EQUIVALENCE
EVALUATIONS, NDA 021123, available at http://www.fda.gov/cder/ob/ (search “Search by Application Number” for “021123”).
11. Roin, supra note 1, at 521 n.86. 12. Id. 13. Kali Labs I, No. 06-CV-3533, 2008 WL 1782283, at *7 (D. N.J. Apr. 17, 2008). 14. Roin also claims that the Flick patent failed to disclose the combination of tramadol and
acetaminophen: “The patent did not claim the tramadol-acetaminophen combination as part of the invention.” Roin, supra note 1, at 520 n.82. While not part of the enumerated claims, the combination was clearly described in Examples 22 and 23 of the Flick patent.
drug candidates are “unpatentable” other than examples such as Ultracet. But Ultracet is a poor example, since it remains patented today, with a patent listed in the FDA Orange Book expiring August 9, 2011.15 Ortho-McNeil’s original patent (the ‘691 patent) was issued in 1994 and reissued on August 1, 2006 (the ‘221 patent) during litigation.16 Thus, the general claim is not supported: prior disclosure in the Flick patent was not a barrier to patenting Ortho-McNeil’s Ultracet.
Perhaps the crux of the claim is narrower—that disclosure resulted in a
lower quality patent. The ‘691 patent certainly attracted more than its share of litigation. Kali Labs filed an ANDA with a “paragraph iv” certification on August 9, 2002, challenging the validity of the ‘691 patent.17 After the 30-month automatic stay expired, the FDA approved the ANDA. Kali Labs was able to register the product with the FDA on April 21, 2005. As the first generic approval, Kali Labs received the 180-day exclusivity period but entered the market “at-risk” since the ‘691 patent litigation was not yet concluded.18
Today, four generic products are registered with the FDA that reference
Ultracet.19 But the ‘691 patent did not prove to be entirely toothless. In July 2007, Kali Labs settled with Ortho-McNeil after the successful reissue of the ‘691 patent (now the ‘221 patent). Under the terms of the settlement, Kali Labs agreed to exit the market by November 15, 2007, paying a lump sum for damages and a royalty-bearing license until withdrawal. Furthermore, the consent agreement upheld the validity of the ‘221 patent. The other generic entrants were more successful at staying in the market, but that result was due to their willingness to challenge the reissued ‘221 patent in litigation. Caraco Pharmaceutical Laboratories filed its ANDA and was sued by Ortho-McNeil in the federal district court in the eastern district of Michigan.20 The
15. U.S. Patent No. 5,336,691 (filed Nov. 10, 1992) (issued Aug. 9, 1994) This patent is now
16. The ‘221 reissued patent retained only the key claim 6 of the ‘691 patent, but added 62 new
additional claims. See Ortho-McNeil Pharmaceutical, Inc. v. Kali Laboratories, Inc., 482 F. Supp. 2d 478, 488 (D. N.J. 2007).
17. Food, Drug, and Cosmetics Act, § 505(j), 21 U.S.C. § 355(j) (2000). By making a
paragraph iv certification, the filer of the ANDA certifies to the FDA that the generic drug will not infringe on existing patents listed in the FDA Orange Book. This certification does not bind the U.S. Patent and Trademark Office, nor does it determine subsequent patent litigation.
18. Market entry was “at risk” because Kali Labs had obtained marketing approval from the
Food and Drug Administration but had not resolved the patent litigation.
19. FDA, Orange Book of Approved Drug Products with Therapeutic Equivalence Evaluations,
NDA 021123, (“FDA NDA 021123”), available at http://www.fda.gov/cder/ob/, (search “Search by Application Number” for “021123”). The four companies are Alpha Pharm, Barr, Caraco, and Kali Labs.
20. Ortho-McNeil Pharm. Inc., v. Caraco Pharm. Labs., Ltd. (Caraco I), No. 04-CV-73698,
2005 WL 2679788 (E.D. Mich. Oct. 19, 2005).
district court held the ‘691 patent was not infringed in October 2005.21 Barr Pharmaceuticals also filed an ANDA and entered the market “at risk.”22 The Federal Circuit ultimately found the ‘221 patent not infringed by Caraco,23 but only after Kali Labs had settled.
C. The Findings of Noninfringement Were Due in Significant Part to the Weight Ratios Chosen by Ortho-McNeil
The facts are considerably more complicated than the “unpatentable”
scenario Roin describes. The Flick patent disclosed a combination of acetaminophen and tramadol for pain relief in a weight ratio of 1:10.24 Ortho-McNeil sought and received FDA approval for Ultracet with a weight ratio of 1:8.67, 37.5 mg of tramadol and 325 mg of acetaminophen.25 In claim six of the ‘691 patent, Ortho-McNeil claimed a weight ratio of “about 1:5.” The Caraco case turned on whether 1:8.67 infringed the claim of “about 1:5.” Based on expert testimony, the District Court construed “about 1:5” to include weight ratios from 1:3.6 to 1:7.1.26 After Caraco amended its ANDA in July 2005 to narrow its manufacturing variability to a minimum of 1:7.5, with a target ratio of 1:8.67, the district court found no infringement.27 The Federal Circuit affirmed on January 19, 2007.28 Apotex received a similar ruling of noninfringement in the Northern District of Illinois in 2007.
Why did Ortho-McNeil claim only “about 1:5” in claim six of the ‘691
patent, instead of a broader claim? Perhaps the presence of the 1:10 disclosure in the Flick patent encouraged them to seek an imprecise distance. When Ortho-McNeil undertook clinical trials for Ultracet, it settled on the 1:8.67 weight ratio on its own accord, and this dosing was the foundation for their New Drug Application.29 Ortho-McNeil could not subsequently change the weight ratios without seeking a new NDA with new clinical trials. In short, their patent problems were largely the result of choices Ortho-McNeil made.
21. Ortho-McNeil Pharm., Inc. v. Kali Labs., Inc. (Kali Labs I), No. 06-CV-3533, 2008 WL
22. Ortho-McNeil Pharm., Inc. v. Kali Labs. (Kali Labs II), No. 02-5705, 2007 WL 1814080, at
23. Ortho-McNeil Pharm., Inc. v. Caraco Pharm. Labs., Ltd. (CaracoII), 476 F.3d 1321, 1329
24. U.S. Patent No. 3,652,589 Example 23 (filed July 27, 1967) (issued Mar. 28, 1972). 25. FDA NDA 021123, supra note 15. 26. Caraco I, No. 04-CV-73698, 2005 WL 2679788, at *4 (E.D. MI. Oct. 19, 2005). 27. Id. at *7 28. Caraco II, 476 F.3d at 1329. 29. FDA NDA 021123, supra note 15, at 10.
D. The Public Domain Did Not Impair Ortho-McNeil’s Commercialization
A key element of Roin’s argument is that the unpatentable nature of
drug candidates in the public domain will prevent commercialization of valuable drugs. Of course that was not true for Ultracet. Ortho-McNeil was in all likelihood aware of the weaknesses of its ‘691 patent, and yet they proceeded to sponsor clinical trials, to complete the New Drug Application, and bring Ultracet to the U.S. market. This does not appear to be a case of surprise or ignorance of the prior publication, as discussed above.
Sometimes a company will proceed with a weaker patent and face the litigation risks. Companies with weaker molecular patents may compensate with aggressive evergreen patenting of uses, combinations, dosages, and extended release formulations, together with non-patent IP strategies such as trade secrecy, data exclusivity, and branding based on trademarks.
We should note the relative position of the ‘691 patent. It does not
claim a patent to the molecules for either acetaminophen or tramadol. McNeil introduced acetaminophen to the U.S. market in 1955 under the brand name Tylenol. The basic patent has long expired, but the brand flourishes through trademark and brand extensions. Ortho-McNeil also introduced tramadol to the U.S. market in 1995 under the brand name Ultram.30 The Flick patent was granted in 1972, and yet generic entry was delayed until 2002 for various reasons. Ultracet could be characterized as an attempt to evergreen 50 mg Ultram by reducing the dose to 37.5 mg and combining it with 325mg of acetaminophen. In the late 1990s, Ultram was listed as one of the top six performing prescription drugs for Ortho-McNeil’s parent, Johnson & Johnson,31 and Tylenol remains a major source of company profits.
In many cases, companies patent drugs even if the molecule is in the
public domain, particularly through method-of-use patents, particular formulations, and combinations. The FDA Orange Book lists many approved drugs with use, formulation, or combination patents. Roin does not explain why these tools are inadequate to bring drugs to market.
30. FDA, Orange Book of Approved Drug Products with Therapeutic Equivalence Evaluations,
NDA 020281, available at http://www.fda.gov/cder/ob/, (search “Search by Application Number” for “020281”) (approved March 3, 1995). I have not been able to find whether the Flick patent was licensed for Ultram.
31. Johnson & Johnson, Annual Report Pursuant to Section 13 of the Securities Exchange Act
of 1934 for the Fiscal Year Ending January 3, 1999 (Form 10-K), at 28 (Apr. 1, 1999).
E. Free Riding Did Not Undermine Ortho-McNeil’s Legitimate Expectations of Return on Investment on Ultracet.
The oft-quoted, but never audited, estimated cost of drug development
is $800 million to 1.2 billion per drug.32 The same research group pegs the cost of clinical trials at $250 million per drug.33 The costs to bring Ultracet to the market were likely much lower than these numbers suggest.
This was not a “needle in a haystack” research project, but a fixed dose
combination of two of the most popular FDA approved analgesics, which were both well-known to Ortho-McNeil. These factors greatly increased the odds of success. Because the separate products were so well known, the FDA approved Ultracet on the basis of a small number of subjects in short-duration (eight-hour) pain studies. The total number of subjects in the universe of submitted studies was 3875; only 1389 patients received Ultracet after randomization.34 The FDA found only three of these studies to be “pivotal,” with only 240 people receiving Ultracet after randomization (1197 total subjects).35 Data exclusivity for FDA purposes would be important for only these three studies. While only the company knows the cost of these studies, very short duration pain studies involving only 1197 subjects might have cost several million dollars, but probably not more than $10 million.
For this investment, Ortho-McNeil reaped significant rewards during
the short effective patent life of Ultracet. In 2006 alone, Ortho-McNeil reportedly sold approximately $350 million of Ultracet.36 Even after patent expiration, a company that has established a strong brand can continue to generate strong sales, despite generic entry, as Ortho-McNeil continues to demonstrate with Tylenol.
F. Ultracet Is Not a Particularly Valuable Drug
At many points, Roin laments the “valuable” or “potentially valuable”
“lost drugs” that companies are ignoring due to unpatentability. There may be excellent examples of socially valuable lost drugs, but Ultracet is not among them. We should first note that both drugs are available separately, and acetaminophen is widely available without a prescription. The key to the FDA approval of Ultracet was its synergistic effects as a fixed-dose
32. Roin, supra note 1, at 510. 33. Id. at 534 n.167. 34. FDA, CENTER FOR DRUG EVALUATION AND RESEARCH, APPLICATION 21-123
http://www.fda.gov/cder/foi/nda/2001/21123_Ultracet_medr_P1.pdf [hereinafter, ULTRACET MEDICAL REVIEW].
35. Id. at 25 tbl.11. 36. Posting of David S. Harper to Orange Book Blog, http://www.orangebookblog.com/2007/
01/caraco_wins_app.html (Jan. 21, 2007). Johnson & Johnson did not release sales figures on Ultracet in its SEC 10-K filings.
combination. In the three pivotal pain studies, Ultracet (TRAM/APAP) was tested against 650 mg acetaminophen (APAP) alone, 75 mg tramadol (TRAM) alone, 400 mg ibuprofen, and a placebo. The trials demonstrated Ultracet as superior to a placebo for dental pain following tooth extraction. In the first hour, acetaminophen was statistically similar to Ultracet, and either therapy was superior to the other options. For hours 2–8, ibuprofen was generally equal to or superior to Ultracet, but both ibuprofen and Ultracet were generally superior to the other treatments.37 The following chart is from one of the pivotal trials discussed in the FDA medical review:38
In short, one would be hard-pressed to say that Ultracet was a breakthrough drug for the treatment of pain, despite the marketing success.
At this point, I fear that I am testing the patience of the readers of a
purportedly “short” Comment, so let us assume, for the sake of argument, that some breakthrough drug candidates may lack ironclad patents. If true, the next question is, How we get these drugs to market? How do we protect first-mover drug companies from unfair generic free riders? Ben Roin proposes data exclusivity periods enforced by the FDA. The next Part examines this potential solution, as well as roads not taken.
37. ULTRACET MEDICAL REVIEW, supra note 30, at 36. On the charts, ibuprofen achieves
greater pain relief after the first hour in all three pivotal studies, but the differences between the treatment groups and the ibuprofen groups were not statistically significant after the first hour.
Roin resists the temptation to tinker with the patent system itself. He
discusses and rejects modifications to the nonobviousness and novelty standards for patents, generally, or even for pharmaceutical patents, specifically.
More attention, however, could have been directed to first-best solutions
to the free rider problem, especially if investments could be protected without deadweight losses. One such example might be direct government funding of clinical trials for these “lost drugs.” When Roin discusses direct government funding, he raises several objections: (1) inability to identify candidates;39 (2) inability to conduct clinical trials;40 (3) congressional reticence to fund the project;41 and (4) inability to market the product.42 The following paragraphs will examine these objections in turn.
In our assumed “lost drug” scenario of breakthrough, important drug
candidates that lack ironclad patent protection, prizes could be offered to private actors to identify these hidden treasures. Indeed, if industry insiders are correct,43 the companies, themselves, reluctantly set these promising compounds aside and would, therefore, be in the best position to re-identify them. Perhaps the size of the reward could vary with the potential health impact of the identified product, addressing two problems simultaneously: information asymmetries in favor of the companies and the significant disconnect between health needs and pharmaceutical innovation.44 The advantages of the patent system are smaller in this scenario since we posit that the valuable drug candidates are already identified, but deliberately cast aside.
As for clinical trials, government funding of clinical trials need not
require the direct employment of civil servants as principal investigators. The government has substantial expertise in administering grants and outsourcing tasks to private entities. The National Institutes of Health extramural research program is but one example. Roin also fails to address a major flaw with private financing of clinical trials: widespread bias in the conduct and dissemination of clinical trial results funded by drug companies. Government funded trials would not be subject to this bias, and we should not assume that other inefficiencies would swamp these benefits.
39. Roin, supra note 1, at 561. 40. Id. at 561–62. 41. Id. at 562–63. 42. Id. at 563–64. 43. Id. at 546–47. 44. For an introduction by proponents to the pharmaceutical prize literature, see James Love &
Tim Hubbard, The Big Idea: Prizes to Stimulate R&D for New Medicines, 82 CHI.-KENT L. REV. 1519 (2007).
Nor should we assume that the federal government is unwilling or
unable to fund these types of studies. The American Recovery and Reinvestment Act appropriated $1.1 billion for government-funded comparative effectiveness trials.45 Perhaps a few “lost drug” demonstration projects could be funded to test whether low-hanging fruit was indeed available before implementing more fundamental changes.
Finally, concerns about marketing seem better founded. If a public
domain drug of sufficient value was identified and proven in clinical trials, perhaps the government should outlicense the product just as a small biotech would choose to stick to its core competencies rather than build a large marketing machine.
Roin concludes that FDA data exclusivity periods are the “best way for
Congress to promote the development of unpatentable drugs . . . .”46 While admirably distinct from the patent system, these extensions would have similar appropriation and incentive effects as reinstating patents on public domain drug candidates.47 These extensions would necessarily be granted without regard to patent doctrines of nonobviousness or novelty, which is another way of saying that a near-infinite variety of existing drugs would be available for patent-like protection via the FDA. Earlier in his article, Roin discussed the dangers of abusive patent strategies,48 but that concern is not adequately addressed for FDA-administered data exclusivity periods. Patent races, rent dissipation, evergreening, and free (cheap) riding would be rampant, so long as the company completed the clinical trials. Rebecca Eisenberg examined the “Problem of New Uses” in 2005 and suggested FDA data exclusivity, but only in exchange for full public disclosure of trial results.49 In short, Roin’s principal contribution here is to extend this conversation, but he is perhaps too hasty to conclude that data exclusivity is the “best way” to commercialize lost drugs.
Assume for the sake of argument that we have breakthrough drugs
languishing in the public domain with weak or inadequate patents. What other proposals address the free rider problem without requiring data exclusivity or patent reform?
Let me suggest one possible approach: charge the free riders a toll to
get on the bus. If a company successfully takes an unpatented drug to FDA
45. American Recovery and Reinvestment Act, Pub. L. No. 111-5, 123 Stat. 115 (Feb. 17,
46. Roin, supra note 1, at 564. 47. Id. at 565. 48. Id. at 557–59. 49. Rebecca S. Eisenberg, The Problem of New Uses, 5 YALE J. HEALTH POL’Y, L. & ETHICS
approval in a New Drug Application, subsequent companies can file ANDAs referencing that drug only if they pay a fee to the company that sponsored the NDA. That fee should be set as a significant multiple of the audited clinical trial costs incurred. Subsequent ANDAs would pay a smaller fee, with some portion going to the first ANDA as a reward for its own first mover status.
This proposal would not necessarily work in the broader economy, but
with prescription drugs, the FDA is the sole gatekeeper of the ANDA process. If the fee multiple is set high enough, the free rider problem is solved, without the distortions introduced by patents or patent-like tools.
In any event, I thank Ben Roin for shedding light on a fascinating area
#4 LAS INICIATIVAS DE ESPAÑA Y MÉXICO PARA COMBATIR TERRORISMO COMPRENSIÓN ORAL 1. Antes de escuchar : a) ¿Ha escuchado usted del movimiento zapatista en México o del grupo juvenil Haïka en España?Utilice el vocabulario siguiente para escribir una definición de ellos: Grupo juvenil – País Vasco español – "cachorros de ETA" – dirigir y fiscalizar – banda
FRUCHTSÄURE- PEELING WAS KANN EIN FRUCHTSÄURE-PEELING WIRKLICH? WIE FUNKTIONIERT ES? UND AUF WELCHE KOMPLIMENTE KANN MAN SICH DANACH FREUEN? WIR HABEN EINEN EXPERTEN UND EINE ANWENDERIN BEFRAGT Wer ohne OP ein paar Fältchen loswerden möchte, hat eine oft beworbene Option: das chemische Peeling. Die nichtinvasive Methode zurVerjüngung der Haut verspricht eine Verbesserung der Hautelastizit�
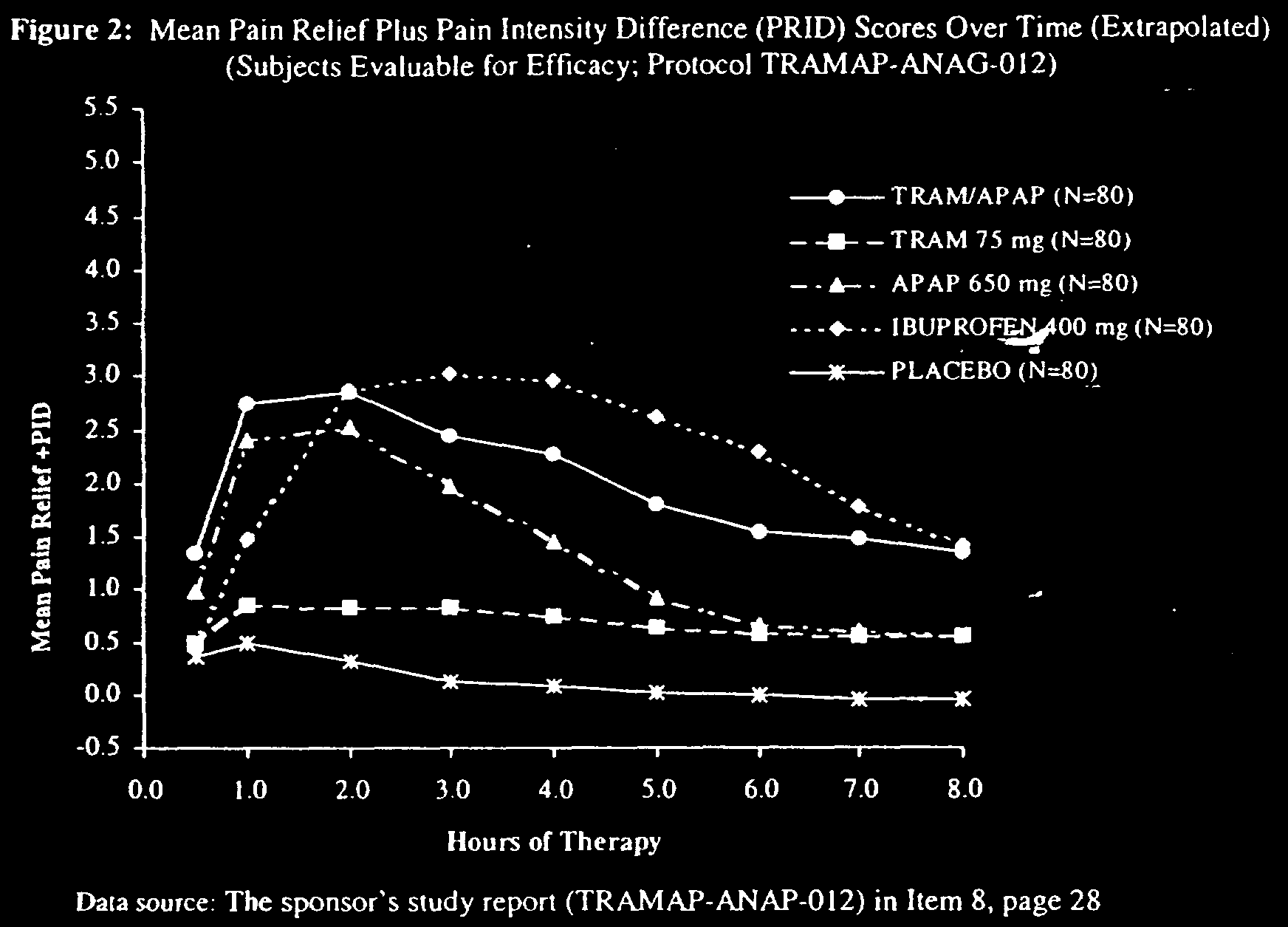 combination. In the three pivotal pain studies, Ultracet (TRAM/APAP) was tested against 650 mg acetaminophen (APAP) alone, 75 mg tramadol (TRAM) alone, 400 mg ibuprofen, and a placebo. The trials demonstrated Ultracet as superior to a placebo for dental pain following tooth extraction. In the first hour, acetaminophen was statistically similar to Ultracet, and either therapy was superior to the other options. For hours 2–8, ibuprofen was generally equal to or superior to Ultracet, but both ibuprofen and Ultracet were generally superior to the other treatments.37 The following chart is from one of the pivotal trials discussed in the FDA medical review:38
In short, one would be hard-pressed to say that Ultracet was a breakthrough drug for the treatment of pain, despite the marketing success.
At this point, I fear that I am testing the patience of the readers of a
purportedly “short” Comment, so let us assume, for the sake of argument, that some breakthrough drug candidates may lack ironclad patents. If true, the next question is, How we get these drugs to market? How do we protect first-mover drug companies from unfair generic free riders? Ben Roin proposes data exclusivity periods enforced by the FDA. The next Part examines this potential solution, as well as roads not taken.
37. ULTRACET MEDICAL REVIEW, supra note 30, at 36. On the charts, ibuprofen achieves
greater pain relief after the first hour in all three pivotal studies, but the differences between the treatment groups and the ibuprofen groups were not statistically significant after the first hour.
Roin resists the temptation to tinker with the patent system itself. He
discusses and rejects modifications to the nonobviousness and novelty standards for patents, generally, or even for pharmaceutical patents, specifically.
More attention, however, could have been directed to first-best solutions
to the free rider problem, especially if investments could be protected without deadweight losses. One such example might be direct government funding of clinical trials for these “lost drugs.” When Roin discusses direct government funding, he raises several objections: (1) inability to identify candidates;39 (2) inability to conduct clinical trials;40 (3) congressional reticence to fund the project;41 and (4) inability to market the product.42 The following paragraphs will examine these objections in turn.
In our assumed “lost drug” scenario of breakthrough, important drug
candidates that lack ironclad patent protection, prizes could be offered to private actors to identify these hidden treasures. Indeed, if industry insiders are correct,43 the companies, themselves, reluctantly set these promising compounds aside and would, therefore, be in the best position to re-identify them. Perhaps the size of the reward could vary with the potential health impact of the identified product, addressing two problems simultaneously: information asymmetries in favor of the companies and the significant disconnect between health needs and pharmaceutical innovation.44 The advantages of the patent system are smaller in this scenario since we posit that the valuable drug candidates are already identified, but deliberately cast aside.
As for clinical trials, government funding of clinical trials need not
require the direct employment of civil servants as principal investigators. The government has substantial expertise in administering grants and outsourcing tasks to private entities. The National Institutes of Health extramural research program is but one example. Roin also fails to address a major flaw with private financing of clinical trials: widespread bias in the conduct and dissemination of clinical trial results funded by drug companies. Government funded trials would not be subject to this bias, and we should not assume that other inefficiencies would swamp these benefits.
39. Roin, supra note 1, at 561. 40. Id. at 561–62. 41. Id. at 562–63. 42. Id. at 563–64. 43. Id. at 546–47. 44. For an introduction by proponents to the pharmaceutical prize literature, see James Love &
Tim Hubbard, The Big Idea: Prizes to Stimulate R&D for New Medicines, 82 CHI.-KENT L. REV. 1519 (2007).
Nor should we assume that the federal government is unwilling or
unable to fund these types of studies. The American Recovery and Reinvestment Act appropriated $1.1 billion for government-funded comparative effectiveness trials.45 Perhaps a few “lost drug” demonstration projects could be funded to test whether low-hanging fruit was indeed available before implementing more fundamental changes.
Finally, concerns about marketing seem better founded. If a public
domain drug of sufficient value was identified and proven in clinical trials, perhaps the government should outlicense the product just as a small biotech would choose to stick to its core competencies rather than build a large marketing machine.
Roin concludes that FDA data exclusivity periods are the “best way for
Congress to promote the development of unpatentable drugs . . . .”46 While admirably distinct from the patent system, these extensions would have similar appropriation and incentive effects as reinstating patents on public domain drug candidates.47 These extensions would necessarily be granted without regard to patent doctrines of nonobviousness or novelty, which is another way of saying that a near-infinite variety of existing drugs would be available for patent-like protection via the FDA. Earlier in his article, Roin discussed the dangers of abusive patent strategies,48 but that concern is not adequately addressed for FDA-administered data exclusivity periods. Patent races, rent dissipation, evergreening, and free (cheap) riding would be rampant, so long as the company completed the clinical trials. Rebecca Eisenberg examined the “Problem of New Uses” in 2005 and suggested FDA data exclusivity, but only in exchange for full public disclosure of trial results.49 In short, Roin’s principal contribution here is to extend this conversation, but he is perhaps too hasty to conclude that data exclusivity is the “best way” to commercialize lost drugs.
Assume for the sake of argument that we have breakthrough drugs
languishing in the public domain with weak or inadequate patents. What other proposals address the free rider problem without requiring data exclusivity or patent reform?
Let me suggest one possible approach: charge the free riders a toll to
get on the bus. If a company successfully takes an unpatented drug to FDA
45. American Recovery and Reinvestment Act, Pub. L. No. 111-5, 123 Stat. 115 (Feb. 17,
46. Roin, supra note 1, at 564. 47. Id. at 565. 48. Id. at 557–59. 49. Rebecca S. Eisenberg, The Problem of New Uses, 5 YALE J. HEALTH POL’Y, L. & ETHICS
approval in a New Drug Application, subsequent companies can file ANDAs referencing that drug only if they pay a fee to the company that sponsored the NDA. That fee should be set as a significant multiple of the audited clinical trial costs incurred. Subsequent ANDAs would pay a smaller fee, with some portion going to the first ANDA as a reward for its own first mover status.
This proposal would not necessarily work in the broader economy, but
with prescription drugs, the FDA is the sole gatekeeper of the ANDA process. If the fee multiple is set high enough, the free rider problem is solved, without the distortions introduced by patents or patent-like tools.
In any event, I thank Ben Roin for shedding light on a fascinating area
combination. In the three pivotal pain studies, Ultracet (TRAM/APAP) was tested against 650 mg acetaminophen (APAP) alone, 75 mg tramadol (TRAM) alone, 400 mg ibuprofen, and a placebo. The trials demonstrated Ultracet as superior to a placebo for dental pain following tooth extraction. In the first hour, acetaminophen was statistically similar to Ultracet, and either therapy was superior to the other options. For hours 2–8, ibuprofen was generally equal to or superior to Ultracet, but both ibuprofen and Ultracet were generally superior to the other treatments.37 The following chart is from one of the pivotal trials discussed in the FDA medical review:38
In short, one would be hard-pressed to say that Ultracet was a breakthrough drug for the treatment of pain, despite the marketing success.
At this point, I fear that I am testing the patience of the readers of a
purportedly “short” Comment, so let us assume, for the sake of argument, that some breakthrough drug candidates may lack ironclad patents. If true, the next question is, How we get these drugs to market? How do we protect first-mover drug companies from unfair generic free riders? Ben Roin proposes data exclusivity periods enforced by the FDA. The next Part examines this potential solution, as well as roads not taken.
37. ULTRACET MEDICAL REVIEW, supra note 30, at 36. On the charts, ibuprofen achieves
greater pain relief after the first hour in all three pivotal studies, but the differences between the treatment groups and the ibuprofen groups were not statistically significant after the first hour.
Roin resists the temptation to tinker with the patent system itself. He
discusses and rejects modifications to the nonobviousness and novelty standards for patents, generally, or even for pharmaceutical patents, specifically.
More attention, however, could have been directed to first-best solutions
to the free rider problem, especially if investments could be protected without deadweight losses. One such example might be direct government funding of clinical trials for these “lost drugs.” When Roin discusses direct government funding, he raises several objections: (1) inability to identify candidates;39 (2) inability to conduct clinical trials;40 (3) congressional reticence to fund the project;41 and (4) inability to market the product.42 The following paragraphs will examine these objections in turn.
In our assumed “lost drug” scenario of breakthrough, important drug
candidates that lack ironclad patent protection, prizes could be offered to private actors to identify these hidden treasures. Indeed, if industry insiders are correct,43 the companies, themselves, reluctantly set these promising compounds aside and would, therefore, be in the best position to re-identify them. Perhaps the size of the reward could vary with the potential health impact of the identified product, addressing two problems simultaneously: information asymmetries in favor of the companies and the significant disconnect between health needs and pharmaceutical innovation.44 The advantages of the patent system are smaller in this scenario since we posit that the valuable drug candidates are already identified, but deliberately cast aside.
As for clinical trials, government funding of clinical trials need not
require the direct employment of civil servants as principal investigators. The government has substantial expertise in administering grants and outsourcing tasks to private entities. The National Institutes of Health extramural research program is but one example. Roin also fails to address a major flaw with private financing of clinical trials: widespread bias in the conduct and dissemination of clinical trial results funded by drug companies. Government funded trials would not be subject to this bias, and we should not assume that other inefficiencies would swamp these benefits.
39. Roin, supra note 1, at 561. 40. Id. at 561–62. 41. Id. at 562–63. 42. Id. at 563–64. 43. Id. at 546–47. 44. For an introduction by proponents to the pharmaceutical prize literature, see James Love &
Tim Hubbard, The Big Idea: Prizes to Stimulate R&D for New Medicines, 82 CHI.-KENT L. REV. 1519 (2007).
Nor should we assume that the federal government is unwilling or
unable to fund these types of studies. The American Recovery and Reinvestment Act appropriated $1.1 billion for government-funded comparative effectiveness trials.45 Perhaps a few “lost drug” demonstration projects could be funded to test whether low-hanging fruit was indeed available before implementing more fundamental changes.
Finally, concerns about marketing seem better founded. If a public
domain drug of sufficient value was identified and proven in clinical trials, perhaps the government should outlicense the product just as a small biotech would choose to stick to its core competencies rather than build a large marketing machine.
Roin concludes that FDA data exclusivity periods are the “best way for
Congress to promote the development of unpatentable drugs . . . .”46 While admirably distinct from the patent system, these extensions would have similar appropriation and incentive effects as reinstating patents on public domain drug candidates.47 These extensions would necessarily be granted without regard to patent doctrines of nonobviousness or novelty, which is another way of saying that a near-infinite variety of existing drugs would be available for patent-like protection via the FDA. Earlier in his article, Roin discussed the dangers of abusive patent strategies,48 but that concern is not adequately addressed for FDA-administered data exclusivity periods. Patent races, rent dissipation, evergreening, and free (cheap) riding would be rampant, so long as the company completed the clinical trials. Rebecca Eisenberg examined the “Problem of New Uses” in 2005 and suggested FDA data exclusivity, but only in exchange for full public disclosure of trial results.49 In short, Roin’s principal contribution here is to extend this conversation, but he is perhaps too hasty to conclude that data exclusivity is the “best way” to commercialize lost drugs.
Assume for the sake of argument that we have breakthrough drugs
languishing in the public domain with weak or inadequate patents. What other proposals address the free rider problem without requiring data exclusivity or patent reform?
Let me suggest one possible approach: charge the free riders a toll to
get on the bus. If a company successfully takes an unpatented drug to FDA
45. American Recovery and Reinvestment Act, Pub. L. No. 111-5, 123 Stat. 115 (Feb. 17,
46. Roin, supra note 1, at 564. 47. Id. at 565. 48. Id. at 557–59. 49. Rebecca S. Eisenberg, The Problem of New Uses, 5 YALE J. HEALTH POL’Y, L. & ETHICS
approval in a New Drug Application, subsequent companies can file ANDAs referencing that drug only if they pay a fee to the company that sponsored the NDA. That fee should be set as a significant multiple of the audited clinical trial costs incurred. Subsequent ANDAs would pay a smaller fee, with some portion going to the first ANDA as a reward for its own first mover status.
This proposal would not necessarily work in the broader economy, but
with prescription drugs, the FDA is the sole gatekeeper of the ANDA process. If the fee multiple is set high enough, the free rider problem is solved, without the distortions introduced by patents or patent-like tools.
In any event, I thank Ben Roin for shedding light on a fascinating area