Levitra has a minimal amount of contraindications which has increased its popularity viagra uk You can buy quality certified medications from us at an affordable price.
Malignehyperthermie.ch
Malignant Hyperthermia—Molecular Testing
Thierry GirardDepartments of Anesthesia and Research, University hospital, Basel, Switzerland
Henrik RueffertDepartment of Anesthesiology and Intensive Care Medicine, University hospital,Leipzig, Germany
Perioperative deaths associated with hyperthermia
have been reported since introduction of general
Malignant hyperthermia (MH) is triggered by all
anesthesia in the 19th century. Only in 1960 did
inhalative anesthetics as well as depolarizing muscle
Denborough and Lovell describe the autosomal domi-
relaxants in genetically predisposed individuals. Pre-
nant mode of inheritance of this potentially fatal disease,
symptomatic testing is important in this potentially
which was then named ‘‘malignant hyperthermia.’’[1]
fatal pharmacogenetic disease. In vitro challenging
Malignant hyperthermia (OMIM 145600) is a classic
of muscle samples with halothane and caffeine is the
pharmacogenetic disorder. Affected individuals are
basis of contracture testing, which until recently was
free of any symptoms in daily life, but in geneti-
the only accepted diagnostic procedure. Contracture
cally predisposed individuals exposure to triggering
testing is invasive and needs an open skeletal muscle
agents causes a dramatic life-threatening increase in
biopsy. Therefore, research in MH focuses on the
development of less invasive procedures. In approxi-mately 85% of MH families, segregation analyses withmicrosatellite markers showed positive linkage of theMH susceptible (MHS) phenotype to the genetic locus
of the skeletal muscle endoplasmatic calcium channel(ryanodine receptor, RYR1). Guidelines for molecular
The clinical symptoms of MH reflect a significantly
genetic diagnosis of MH were recently published and
accelerated cell metabolism in the skeletal muscles,
allow for molecular genetic diagnosis of MH suscep-
which is initiated by certain triggering agents, i.e., all
tibility in case of identification of a MH causative
halogenated inhalative anesthetics and the depolariz-
mutation. Because of the locus and allelic heterogene-
ing muscle relaxant succinylcholine. The elevated mus-
ity of MH, absence of MH associated mutations do
cle activity leads to increases in skeletal muscle tone
not allow for negative MH diagnosis.
(masseter spasm, general rigidity) and later to signsof muscular damage such as elevated creatine kinase,hyperkaliemia, myoglobinemia, and myoglobinuria. Cellular and systemic hypermetabolism present with
combined metabolic and respiratory acidosis as wellas an elevation in body temperature and finally result
Malignant hyperthermia is a classic pharmacogenetic
in multiorgan failure. During MH episodes, not all of
disease. Apparently, normal individuals with a genetic
the classic symptoms have to be present. Malignant
predisposition to MH exhibit a potentially lethal
hyperthermia might also present with a single symp-
increase in metabolism following contact with trigger-
tom only or symptoms only marginally outside a
ing agents, i.e., all halogenated inhalative anesthetics
normal range. These abortive cases make the diagnosis
and the depolarizing muscle relaxant succinylcholine.
of MH on the basis of clinical presentation difficult
This article briefly describes the clinical symptoms
and only full-blown MH episodes can be accurately
and therapy of MH before giving insight into the
classified as true MH events. Early recognition and
pathophysiology and related genetics. Traditional
treatment in case of suspicion of a MH reaction influ-
phenotyping methods that use contracture testing as
ences the outcome. Monitoring during anesthesia has
well as the recently introduced guidelines for molecular
substantially changed in the last 20 years. Continuous
genetic testing are discussed. The goal of this article
measurement of carbon dioxide production, as well as
is to elucidate the importance of presymptomatic test-
arterial oxygen saturation together with improved
ing for MH and the impact of molecular genetic
alertness of the anesthesiologist towards MH, all
considerably reduced the likelihood of full-blown
Encyclopedia of Medical Genomics and Proteomics DOI: 10.1081/E-EDGP-120040294Copyright # 2005 by Taylor & Francis. All rights reserved.
Malignant Hyperthermia—Molecular Testing
MH episodes. Supportive measures and specific
weakness of the proximal limb.[11] Skeletal defects
therapy are essential in case of a MH episode. Trigger
including congential hip dislocation, thoracic defor-
agents have to be discontinued followed by ventilation
mities, pes cavus, clubfoot, and kyphoscoliosis are
of the patient with pure oxygen and physical body
associated with CCD. Like MH, CCD has been
cooling. Specific therapy consists of the calcium chan-
linked to the locus of RYR1. Central core disease is
nel blocker dantrolene, which has to be immediately
the only myopathy being closely associated with MH
administered. With these therapeutic interventions,
the mortality of MH has decreased dramatically from
Because of its multitude of associated RYR muta-
70% to less than 10%.[2] The importance of dantrolene
tions, MH shows considerable allelic heterogeneity;
in the therapy of MH episodes makes it mandatory to
locus heterogeneity has also been described. Besides
have this drug available in every anesthetic institution
chromosome 19, additional genetic loci on chromo-
somes 1q32, 3q13.1, 5q, 17q11.2, 7q.23–q21.1 have
The incidence of MH is difficult to determine.
been linked to MH.[12] However, for most of these loci,
Clinical data probably underestimate the true genetic
no candidate gene has yet been identified, except for
predisposition, because the phenotypic presentation
chromosomes 1q23 and 7q23–q21.1. Here, the genes
after contact with triggering agents is highly variable
encoding for the alpha1- and alpha2=delta subunit
and many full-blown episodes occurred in patients
of the DHPR could be detected. In MHS patients,
who had previously undergone several uneventful
Monnier et al.[13] found a mutation in the gene encod-
anesthesias despite having contact with trigger agents.
ing for the alpha1-subunit of DHPR (CACNA1S).
Former estimates of about 1 : 15,000 anesthetics in
Furthermore, results of an extended transmission dis-
children and about 1 : 50,000 in adults might well
equilibrium test (ETDT) on European MH families
suggested that multiple interacting genes influence theMH phenotype while the RYR1 gene on chromosome19 represents the major locus for MH susceptibility.[14]
The animal model of MH, the porcine stress syndrome,
allowed for a deeper insight into the pathophysiologyof MH. Compared with MH negative (MHN) pigs,
Specific testing must be used to diagnose MH suscep-
skeletal muscle strips from MHS pigs showed an
tibility, as MH is a subclinical myopathy. The aim of
increase in myoplasmic calcium concentration if chal-
testing for MH is to establish the MH status of
lenged with triggering agents.[3] Myoplasmic calcium
1) patients who had a possible MH episode and
concentration is essentially regulated by the calcium
2) relatives of patients already tested and found to be
release channel of the sarcoplasmic reticulum: the
MHS. Presymptomatic testing for MH confirms or
skeletal muscle isoform of the RYR1. Moreover, the
excludes MH susceptibility before the administration
intracellular Ca2þ release is finely co-ordinated with
of trigger agents. This is important, because safe alter-
the voltage dependent dihydropyridine (DHPR) recep-
natives exist for anesthesia in patients known to be
tor of the transverse tubule interacting with RYR1.
MHS. Regional anesthesia with any local anesthetic
Linkage analyses revealed RYR1 as the primary locus
and total intravenous anesthesia without the use of
for MH susceptibility.[4,5] The gene encoding for
depolarizing neuromuscular blocking drugs is safe for
RYR1 is located on chromosome 19q13.1. Ryanodine
MHS patients.[2] The gold standard for determining a
receptor channel is a homotetramer and represents one
predisposition for MH is an in vitro muscle contrac-
of the largest proteins in the human body. Each sub-
ture test. After an open muscle biopsy, fresh human
unit consists of 5038 amino acids, encoded by a genomic
skeletal muscle is challenged in vitro with caffeine
DNA of 160 kb and 106 exons.[6] In pigs, a mutation at
and halothane. Contractile thresholds to both drugs
nucleotide position 1843, substituting a thymine for
are determined in several muscle strips. Standardized
a cytosine is responsible for MH in an autosomal
protocols for the European in vitro contracture test
recessive mode of inheritance.[7] The same mutation
(IVCT) and the North American caffeine–halothane
was found in humans at position 1840, but in contrast
contracture test (CHCT) have been published.[15,16]
to pigs the MH disposition is dominantly inherited.
Although the two protocols slightly differ, they both
Up to 85% of MH families show linkage to the locus
yield reproducible and comparable results.
of RYR1.[8] Today more than 40 mutations in the
Because MH is a potentially fatal disease, any
RYR1 gene are associated with MH and=or central
diagnostic test requires a high sensitivity in order to
core disease (CCD).[9,10] Central core disease is an
keep the risk of false-negative MH diagnoses as low
autosomal dominant congential myopathy, clinically
as possible.[17] In case of a wrong MHN diagnosis,
characterized by a slow or nonprogressive muscle
the application of MH triggering agents could be
Malignant Hyperthermia—Molecular Testing
detrimental. The sensitivity and specificity of the IVCT
Mutations of the gene encoding the ryanodine
receptor type 1 gene included in guidelines for molecular
Since an invasive open muscle biopsy is required for
genetic diagnosis of malignant hyperthermia (MH)
contracture testing, MH research has focused on the
establishment of noninvasive or less invasive testing
for MH susceptibility. With the introduction of mole-
cular genetic methods and the identification of a single
mutation responsible for the porcine stress syndrome,
many anesthesiologists hoped that finally noninvasive
testing for MH would soon be available. However,allelic and locus heterogeneity of MH has diminished
this expectation. Recently, an important step towards
less invasive MH testing has been made by the
European Malignant Hyperthermia Group (EMHG).
The group published guidelines for molecular genetic
diagnosis of MH susceptibility in 2001.[18] Fifteen
RYR1 mutations were included in these guidelines,all of which had been proven to be causative through
functional analyses. This list of mutations approved
for molecular MH diagnoses is constantly up-
dated, and since publication of the guidelines, another
http:==www.emhg.org). As mentioned above, any
diagnostic approach to MH has to accomplish thehighest possible sensitivity. Incorrect negative MH
diagnoses have to be prevented. This is important
not only for the individuals who are tested, but also
for their offspring. Therefore, the EMHG guidelines
for molecular genetic testing in MH do not allow for
negative MH diagnosis exclusively on the basis of
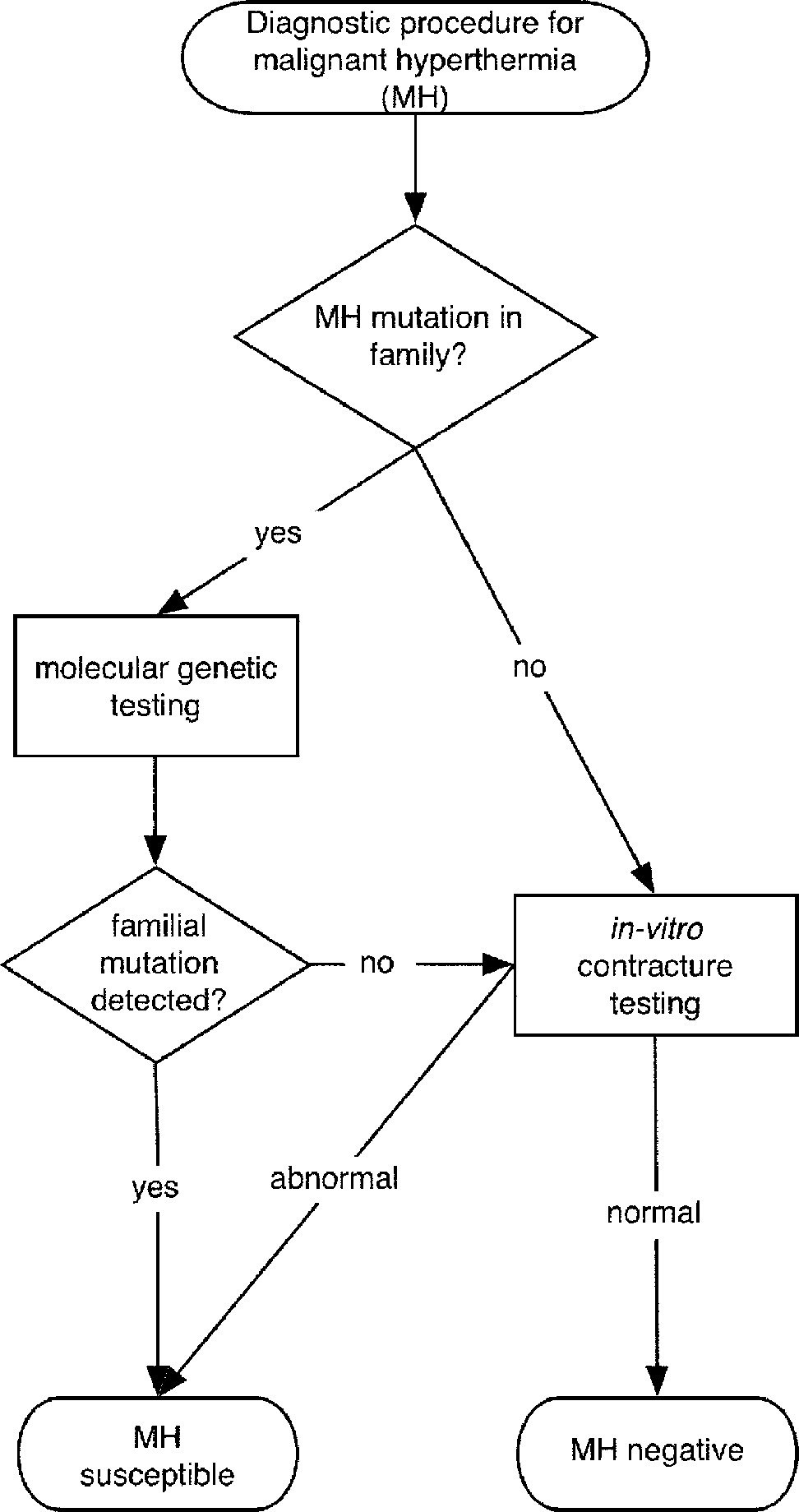
genetic investigations. Following the EMHG guide-lines, first-degree relatives of individuals carrying a
MH causative mutation are investigated for this
familial mutation. While mutation carriers can be
diagnosed MHS, absence of the familial mutation does
EMHG, included in the guidelines of the European MH Group;
not allow for a MHN diagnosis (Fig. 1). The latter sub-
NAMHG, included in the North American MH Group.
jects subsequently require an open muscle biopsyfollowed by contracture testing in order to have theirMH status established.[18] A similar proposal was
is cost-effective if performed in selected individuals.
made by the North American MH Group, suggesting
Total costs of contracture testing is estimated to be
17 MH associated mutations in the RYR1 gene
at U.S.$ 5000–6000[9] and although charges for genetic
(Table 1).[9] Knowledge of mutation frequency for
investigations are not yet defined in every country, they
centers performing molecular genetic investigations
can be expected to be considerably less. Considering
in MH is necessary in order to reduce the number of
the fact that contracture testing involves an open
mutations screened for. For several countries, such
muscle biopsy and can only be performed at specific
investigations were determined and found to be geogra-
centers, the advantages of molecular testing are
phically different.[19] Therefore, it appears to be useful
obvious from the patients’ point of view.
to screen index patients of MH families for the most
Molecular genetic diagnosis of MH is challenged by
frequent MH mutations of their region. Once a muta-
reports of discordances between the genotype and
tion is identified in a family, then additional relatives
phenotypes determined by IVCT,[20] as well as by
can be investigated by molecular methods. If detailed
spontaneous occurrences of MH.[21] We have so
pedigree information is available and individuals are
far performed genetic analyses in 67 individuals, 32
carefully selected, then a 50% rate of positive molecu-
of them were found to be MHS. Of the remaining
lar investigations is to be expected. Even though
35, 20 genetically negative persons underwent IVCT
patients with negative genetic results have to undergo
and all but one were MHN.[19] This calculates to
both, genetic and contracture testing, molecular testing
a negative predictive value of 0.95 (95% confidence
Malignant Hyperthermia—Molecular Testing
The heterogenetic nature of the disease leads to twoimportant drawbacks: 1) Patients not carrying thefamilial mutation must still undergo contracturetesting and 2) only 50% of MH families have knownmutations, some of which are not yet classified as beingcausative. Research in MH will continue to focus onthe aim to offer less invasive MH testing for as manypatients as possible. This involves screening for novelmutations followed by characterization using func-tional analyses. In addition, research also continuesto focus on alternative diagnostic procedures. Severalresearch groups have published preliminary resultsusing either Epstein-Barr Virus (EBV)-immortalizedb-lymphocytes, primary cultures of human skeletalmuscle cells or in vivo microinjections of trigger agents.
In the future, increased knowledge of causative MH
mutations and the development of molecular genetictechniques and gene chip technology might lead to pre-operative genetic analysis and risk profiles. This wouldcertainly increase perioperative patient safety.
We would like to thank Mrs. Joan Etlinger, B.A.,Scientific
1. Denborough, M.; Lovell, R.R. Anaesthetic deaths
2. Gronert, G.; Antonigni, J.; Pessah, I. Malignant
hyperthermia. In Anesthesia, 5th Ed.; Miller, R.,
Fig. 1 Diagram of the diagnostic procedures for malignant
Ed.; Churchill Livingstone: New York, 2000;
3. Iaizzo, P.A.; Klein, W.; Lehmann-Horn, F.
interval 0.75–0.99) for genetic testing and thus, the
Fura-2 detected myoplasmic calcium and its cor-
conservative approach of the EMHG guidelines seems
relation with contracture force in skeletal muscle
to be justified. Today molecular testing for MH is an
from normal and malignant hyperthermia suscep-
important progress and clearly advantageous for the
tible pigs. Pflugers Arch. 1988, 411 (6), 648–653.
patients. Contracture testing and molecular genetics,
4. McCarthy, T.V.; Healy, J.M.; Heffron, J.J.;
the two pillars of MH diagnosis are complementary
Lehane, M.; Deufel, T.; Lehmann-Horn, F.;
Farrall, M.; Johnson, K. Localization of themalignant hyperthermia susceptibility locus tohuman chromosome 19q12–13.2. Nature 1990,343 (6258), 562–564.
5. MacLennan, D.H.; Duff, C.; Zorzato, F.; Fujii, J.;
Phillips, M.; Korneluk, R.G.; Frodis, W.; Britt,
The introduction of the guidelines for molecular
B.A.; Worton, R.G. Ryanodine receptor gene
genetic diagnosis of MH susceptibility is an important
is a candidate for predisposition to malignant
hyperthermia. Nature 1990, 343 (6258), 559–561.
Malignant Hyperthermia—Molecular Testing
6. Phillips, M.S.; Fujii, J.; Khanna, V.K.; DeLeon,
voltage-dependent calcium-channel receptor in
S.; Yokobata, K.; de Jong, P.J.; MacLennan,
skeletal muscle. Am. J. Hum. Genet. 1997, 60 (6),
D.H. The structural organization of the human
skeletal muscle ryanodine receptor (RYR1) gene.
14. Robinson, R.; Hopkins, P.; Carsana, A.; Gilly,
7. Fujii, J.; Otsu, K.; Zorzato, F.; de Leon, S.;
Jurkat-Rott, K.; Muller, C.; Shaw, M.A. Several
MacLennan, D.H. Identification of a mutation
hyperthermia phenotype. Hum. Genet. 2003,
in porcine ryanodine receptor associated with
malignant hyperthermia. Science 1991, 253 (5018),
15. European Malignant Hyperpyrexia Group. A
protocol for the investigation of malignant hyper-
8. Robinson, R.; Curran, J.L.; Hall, W.J.; Halsall,
pyrexia (MH) susceptibility. Br. J. Anaesth. 1984,
P.J.; Hopkins, P.M.; Markham, A.F.; Stewart,
A.D.; West, S.P.; Ellis, F.R. Genetic heterogeneity
16. Larach, M.G. Standardization of the caffeine
and HOMOG analysis in British malignant hyper-
thermia families. J. Med. Genet. 1998, 35 (3),
Anesth. Analg. 1989, 69 (4), 511–515.
9. Nelson, T.E.; Rosenberg, H.; Muldoon, S.M.
17. Allen, G.C.; Brubaker, C.L. Human malignant
Genetic testing for malignant hyperthermia in
hyperthermia associated with desflurane anesthe-
North America. Anesthesiology 2004, 100 (2),
sia. Anesth. Analg. 1998, 86 (6), 1328–1331.
18. Urwyler, A.; Deufel, T.; McCarthy, T.; West, S.
10. McCarthy, T.V.; Quane, K.A.; Lynch, P.J. Rya-
Guidelines for molecular genetic detection of
nodine receptor mutations in malignant hyper-
susceptibility to malignant hyperthermia. Br. J.
thermia and central core disease. Hum. Mutat.
19. Girard, T.; Treves, S.; Voronkov, E.; Siegemund,
11. Tilgen, N.; Zorzato, F.; Halliger-Keller, B.;
M.; Urwyler, A. Molecular genetic testing for
malignant hyperthermia susceptibility. Anesthe-
Schneider, C.; Hauser, E.; Lehmann-Horn, F.;
Muller, C.R.; Treves, S. Identification of four
20. Robinson, R.L.; Anetseder, M.J.; Brancadoro, V.;
novel mutations in the C-terminal membrane
Van Broekhoven, C.; Carsana, A.; Censier, K.;
spanning domain of the ryanodine receptor 1:
Fortunato, G.; Girard, T.; Heytens, L.; Hopkins,
association with central core disease and altera-
P.M.; Jurkat-Rott, K.; Klinger, W.; Kozak-
tion of calcium homeostasis. Hum. Mol. Genet.
Ribbens, G.; Krivosic, R.; Monnier, N.; Nivoche,
Y.; Olthoff, D.; Rueffert, H.; Sorrentino, V.;
12. Jurkat-Rott, K.; McCarthy, T.; Lehmann-Horn,
Tegazzin, V.; Mueller, C.R. Recent advances in
F. Genetics and pathogenesis of malignant
the diagnosis of malignant hyperthermia suscep-
tibility: how confident can we be of genetic
testing? Eur. J. Hum. Genet. 2003, 11 (4), 342–348.
13. Monnier, N.; Procaccio, V.; Stieglitz, P.; Lunardi,
21. Rueffert, H.; Olthoff, D.; Deutrich, C. Sponta-
J. Malignant-hyperthermia susceptibility is asso-
neous occurrence of the disposition to malignant
ciated with a mutation of the alpha 1-subunit
hyperthermia. Anesthesiology 2004, 100 (3),
of the human dihydropyridine-sensitive L-type
OLDHAM GL16 - Li-ionSAFETY LAMP GL16 CAPLAMP - FEATURES & BENEFITS: Extreme lightweight of the Lithium-ion battery: Reduced weight for user to carry, less fatigue Lithium-ion battery does not suffer from “memory effect”: Full capacity available every time after recharge Lithium-ion battery has low self-discharge rate: Long shelf life if stored during mine down-time Ru
Poster Session IV Wednesday, June 20 Presenter’s name is in bold and is subject to change. electric field. In particular, cell displacement rate was higher for cells culturedonto hydrogel substrate and myotubes contraction rate increased as a conse- THE ROLE OF EPHB/EPHRIN-B INTERACTIONS IN CELL quence of the frequency increasing. This frequency-dependent response of ATTACHMENT AND
 Malignant Hyperthermia—Molecular Testing
The heterogenetic nature of the disease leads to twoimportant drawbacks: 1) Patients not carrying thefamilial mutation must still undergo contracturetesting and 2) only 50% of MH families have knownmutations, some of which are not yet classified as beingcausative. Research in MH will continue to focus onthe aim to offer less invasive MH testing for as manypatients as possible. This involves screening for novelmutations followed by characterization using func-tional analyses. In addition, research also continuesto focus on alternative diagnostic procedures. Severalresearch groups have published preliminary resultsusing either Epstein-Barr Virus (EBV)-immortalizedb-lymphocytes, primary cultures of human skeletalmuscle cells or in vivo microinjections of trigger agents.
Malignant Hyperthermia—Molecular Testing
The heterogenetic nature of the disease leads to twoimportant drawbacks: 1) Patients not carrying thefamilial mutation must still undergo contracturetesting and 2) only 50% of MH families have knownmutations, some of which are not yet classified as beingcausative. Research in MH will continue to focus onthe aim to offer less invasive MH testing for as manypatients as possible. This involves screening for novelmutations followed by characterization using func-tional analyses. In addition, research also continuesto focus on alternative diagnostic procedures. Severalresearch groups have published preliminary resultsusing either Epstein-Barr Virus (EBV)-immortalizedb-lymphocytes, primary cultures of human skeletalmuscle cells or in vivo microinjections of trigger agents.