Levitra has a minimal amount of contraindications which has increased its popularity levitra uk You can buy quality certified medications from us at an affordable price.
Ftp.bork.embl-heidelberg.de
Linkage limits the power of natural selection in Drosophila Andrea J. Betancourt*† and Daven C. Presgraves†
Department of Biology, University of Rochester, Rochester, NY 14627
Edited by M. T. Clegg, University of California, Riverside, CA, and approved August 26, 2002 (received for review May 8, 2002)
Population genetic theory shows that the efficacy of natural
First, we ask whether protein evolution is constrained by linkage. selection is limited by linkage—selection at one site interferes with
Second, we ask whether the efficacy of weak selection is com-
selection at linked sites. Such interference slows adaptation in
promised by a stream of strongly selected traffic at nearby sites. asexual genomes and may explain the evolutionary advantage of sex. Here, we test for two signatures of constraint caused by linkage in a sexual genome, by using sequence data from 255 The Data. Coding sequences from 102 D. melanogaster and D. Drosophila melanogaster and Drosophila simulans loci. We find simulans genes were downloaded from GenBank, and aligned by
that (i) the rate of protein adaptation is reduced in regions of low
eye using SE-AL V. 1.0. For genes with multiple transcripts of
recombination, and (ii) evolution at strongly selected amino acid
different lengths, we used the longest transcript; for those with
sites interferes with optimal codon usage at weakly selected,
multiple transcripts of the same length, we used an arbitrarily
tightly linked synonymous sites. Together these findings suggest
selected transcript. The remaining 153 genes (for which W. that linkage limits the rate and degree of adaptation even in
Swanson generously provided alignments) come from a D.recombining genomes. simulans male-specific EST screen (20) and their D. melanogas-ter homologues, downloaded from Flybase (http:͞͞flybase.bio. Natural selection is imperfect. To become fixed, beneficial indiana.edu).WeusedonlycodingESTsfromthisscreenthatwere
mutations must overcome both stochastic loss and inter-
either nonredundant or that were the most rapidly evolving of a set
ference from selection at linked loci. In asexual genomes, where
of redundant ESTs. Although ESTs are unreplicated single-pass
linkage is complete, two kinds of interference compromise
sequences, none of the results in this paper are caused by a
adaptation. The first, ‘‘ruby-in-the-rubbish’’ interference, occurs
difference in sequencing error rate (and consequent elevated
because beneficial mutations often appear on genetic back-
divergence estimates) between the EST and reference quality data,
grounds loaded with segregating deleterious mutations. Since
as all of our results are also found within the EST data set.
deleterious mutations are, on average, probably more strongly
selected than favorable ones, adaptation is mostly limited to
Estimates of Divergence, Recombination, and Codon Usage. We used
those lucky few beneficial mutations that arise on unloaded
maximum likelihood estimates of the rates of amino acid (dN)
backgrounds (1–4). The second form of interference, ‘‘clonal’’
and synonymous (dS) site divergence using PAML (22). One
interference, is caused by competition among multiple segre-
anomalously high dS value (1.71) was excluded from the analysis
gating beneficial mutations (1, 5–7). Because only one asexual
(excluding this value does not affect the results). We used a
genome can be fixed at a time, adaptive substitutions are forced
likelihood ratio test of Goldman and Yang (23) to test for a
to be nearly sequential. Both kinds of interference limit the rate
significant excess of amino acid substitutions in those genes with
dN͞dS Ͼ 1. Briefly, we used PAML to calculate the likelihood
The effects of both kinds of interference can be thought of as
under the null model of equal dN and dS (L0) and the alternative
a reduction in the effective population size (Ne) caused by
model with dN and dS free to vary (L1). The likelihood test
selection at linked loci (11–13). Recombination, by alleviating
statistic (Ϫ2[ln L0 Ϫ ln L1]) was then compared with the 2
interference between linked sites, alleviates this reduction in Ne.
distribution with 1 degree of freedom. dN͞dS Ͼ 1 is strong
Consequently, genomic regions that differ in recombination rate
evidence for adaptive evolution, as this test is conservative.
also differ in effective size—indeed, this is the basis of the
We estimated GC-content and the frequency of optimal codon
well-known correlation between recombination rate and levels
usage (Fop) by using the online program CODONW (http:͞͞
of neutral polymorphism (4Ne times the neutral mutation rate)
bioweb.pasteur.fr͞seqanal͞interfaces͞codonw.html). Fop is the
seen in the genomes of Drosophila, humans, and others (14–18).
proportion of codons in a gene that are optimal codons, defined
That variation in linkage affects levels of neutral polymorphism
as those used most frequently in highly expressed genes (24).
suggests that it may also affect rates of nonneutral substitution.
Optimal codons for both species were assumed to be those of D.
In particular, adaptive evolution may be limited in regions of low
melanogaster. Although we estimate Fop from a single sequence
recombination (i.e., where Ne is reduced) or in situations of
from each species, these estimates should accurately reflect
extreme linkage (e.g., among sites within the same gene).
population levels of optimal codon usage (25).
Here we ask whether linkage systematically constrains adap-
We estimated recombination rates by using the data and
tation in the Drosophila genome. We use divergence estimates
standard method of Kliman and Hey (26). For the X, second, and
from 255 Drosophila melanogaster and Drosophila simulans loci.
third chromosomes, we fit least-squares polynomial curves re-
These data are unique in that they include a large number of
lating recombination rate to DNA content per interval on the
rapidly evolving genes, many of which are candidate male
cytological map (all curves have R2 Ͼ 0.989), and used equations
accessory gland proteins (Acps) and thus likely targets of sexual
from these curves (available upon request) to predict recombi-
selection (19–21). We have reason to believe a priori not only
nation rates. Estimates for X-linked loci were multiplied by 4͞3
that many of these substitutions are adaptive, but also that many
to correct for the absence of recombination in Drosophila males.
of these genes may have experienced a long history of sexual
selection possibly predating the D. melanogaster–D. simulans
split. Such genes are especially good candidates for detecting the
This paper was submitted directly (Track II) to the PNAS office.
signature of interference. We therefore use these data to test for
*To whom correspondence should be addressed. E-mail: [email protected].
two kinds of limits imposed on adaptive evolution by linkage.
†A.J.B. and D.C.P. contributed equally to this work. 13616 –13620 ͉ PNAS ͉ October 15, 2002 ͉ vol. 99 ͉ no. 21
www.pnas.org͞cgi͞doi͞10.1073͞pnas.212277199
For the Y and dot-fourth chromosomes, we assume recombina-
tion is absent. The mean recombination rate in this data set is c ϭ
0.0029 centimorgan (cM)͞kb, close to the global mean for the D.melanogaster genome (see supplimentary information for ref.
27). We therefore defined ‘‘low’’ recombination as c Ͻ 0.0029
and ‘‘high’’ as c Ͼ 0.0029. We used recombination rate estimates
from D. melanogaster for both species, as recombination data for
D. simulans are sparse. Although recombination rates in D.simulans may differ (they are, on average, Ϸ30% higher than in
D. melanogaster; see ref. 28), subtle local changes in recombi-
nation rate are unlikely to lead to misclassification of loci into
high vs. low recombination regions. Changes in recombination
rate that do result in misclassification likely contribute noise to
our analysis, but this would obscure rather than create the
Statistical Tests. For nonnormally distributed variables, we used
nonparametric Spearman rank correlations to test for associa-
tions and permutation t tests (with null distributions generated
by Ն10,000 randomization of the data) to test for differences
between means. For multivariate analyses, nonnormal variables
were first normalized by log-transformation. All tests are two-
tailed. Means are reported with Ϯ 1 SE. All data are available
online in Table 1, which is published as supporting information
Results and Discussion Does Linkage Limit Protein Evolution? We first ask whether linkage
limits protein adaptation. Rates of adaptive protein evolution, if
limited by interference, should be relatively constrained in
regions of low versus high recombination. We therefore test for
an excess (paucity) of rapid evolution in regions of high (low)
recombination. It is important to note, however, that slowly
evolving genes (those mostly subject to purifying selection)
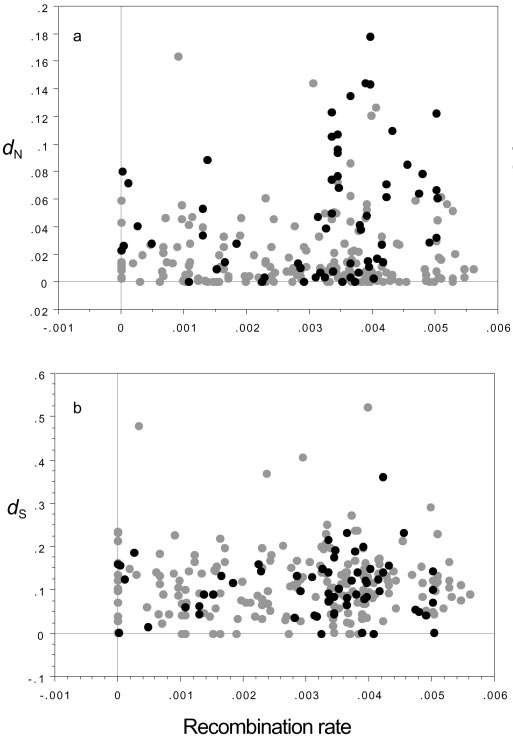
should occur in all recombinational environments. A plot of dN
vs. recombination should thus reveal a wedge-shaped distribu-
tion, with slowly evolving genes in regions of low recombination
and both slowly and rapidly evolving genes in regions of high
N, the rate of amino acid substitution (a), and dS, the rate of silent
substitution (b), vs. recombination rate (in cM͞kb). Black and gray circles are
recombination. Fig. 1a confirms this prediction; genes in high
putative Acp’s and non-Acp’s, respectively.
recombination environments show both a higher mean and
variance of dN values than those in low recombination environ-
ments (dN,high ϭ 0.031 Ϯ 0.003; dN,low ϭ 0.019 Ϯ 0.002; t test ϭ
data (24.4%), our results do not depend on some peculiarity of
2.780, P ϭ 0.007, F151,103 ϭ 2.524, P Ͻ 0.0001; a qualitatively
these genes other than their rapid evolution. Not surprisingly, if
similar pattern appears in a plot of dN͞dS vs. recombination rate,
all candidate Acp’s are excluded from the analysis, too few
rapidly evolving genes remain to detect a pattern. But within
We can eliminate three alternative explanations for this
Acp’s, rapid protein evolution is also largely confined to regions
regional difference in evolutionary rates. First, this wedge pat-
of high recombination (dN,high ϭ 0.057 Ϯ 0.007; dN,low ϭ 0.029 Ϯ
tern is not caused by regional differences in mutational input (as
0.007; t test ϭ 2.369, P ϭ 0.017; F44,18 ϭ 2.884, P ϭ 0.013; dS,high
might be seen, e.g., if recombination were mutagenic) as such
ϭ 0.111 Ϯ 0.011; dS,low ϭ 0.101 Ϯ 0.013; t test ϭ 0.514, P ϭ 0.601;
differences would produce a similar pattern for dS. The distri-
F44,18 ϭ 1.800, P ϭ 0.155). We therefore conclude that rates of
EVOLUTION
S is not, however, wedge-shaped; neither the means
protein adaptation are constrained in the low recombination
nor the variances differ between regions of high and low
environments of the Drosophila genome, as predicted by popu-
recombination (Fig. 1b; dS,high ϭ 0.118 Ϯ 0.006, dS,low ϭ 0.108 Ϯ
0.007; t test ϭ 1.081, P ϭ 0.282; F150,103 ϭ 1.012, P ϭ 0.947).
Second, this result is also inconsistent with most rapid evolution
Does Rapid Protein Evolution Limit Weak Selection at Linked Sites?
being caused by the fixation of many slightly deleterious muta-
We now turn to a second test of the effect of linkage on
tions. In that case, we would expect higher rates of evolution in
adaptation. We ask whether evolution at strongly selected
regions of low recombination (where Ne is reduced by interfer-
(amino acid) sites limits the efficacy of selection at tightly linked,
ence). Other studies have noted such elevated rates of protein
weakly selected (synonymous) sites. Synonymous sites are not
divergence in low recombination regions, likely because, in
selectively equivalent, as certain synonymous codons (‘‘optimal’’
contrast to our data set, most of the genes in these studies are
or ‘‘preferred’’ codons) are used more frequently than others,
relatively conserved and therefore subject mainly to purifying
apparently because of selection for translational efficiency and
selection (29–32). Although many of the genes in this study are
accuracy (33–36). But selection coefficients acting on alternative
also relatively conserved, the signal of interference on adaptive
synonymous codons are on the order of 1͞Ne, i.e., approaching
evolution detected here (elevated dN in high recombination
the limits of selection (36). Such weakly selected sites are
regions) is detectable despite the opposing signal from relaxed
especially susceptible to interference from selection at linked
purifying selection (elevated dN in low recombination regions).
sites (11, 37–39). Two particularly relevant studies (40, 41) have
Third, although candidate Acp’s constitute a large fraction of the
shown, using different models, that a series of strongly selected
PNAS ͉ October 15, 2002 ͉ vol. 99 ͉ no. 21 ͉ 13617
that this is indeed the case. As the rate of amino acid substitution
increases, third position GC-content decreases significantly (dN,
D. melanogaster: rs ϭ Ϫ0.540, P Ͻ 0.0001; dN, D. simulans: rs ϭ
Ϫ0.549, P Ͻ 0.0001; dN͞dS, D. melanogaster: rs ϭ Ϫ0.569, P Ͻ
0.0001; dN͞dS, D. simulans: rs ϭ Ϫ0.584, P Ͻ 0.0001).
Because Acp’s as a class show both low optimal codon usage
(19) and rapid protein evolution (19, 20), it is possible that the
above findings are entirely due to some special property of these
genes. We can rule out this possibility, however. The correlation
between dN and optimal codon usage remains even when Acp’s
are excluded from the analysis (D. melanogaster: rs ϭ Ϫ0.452, P Ͻ
0.0001; D. simulans: rs ϭ Ϫ0.431, P Ͻ 0.0001). Moreover, the
correlation exists, and is in fact stronger, within Acp’s (D.melanogaster: rs ϭ Ϫ0.695, P Ͻ 0.0001; D. simulans: rs ϭ Ϫ0.711,
P Ͻ 0.0001). This stronger correlation probably reflects the fact
that other factors known to contribute to variation in optimal
codon usage—tissue specificity, gene expression level, and gene
length (reviewed in refs. 34 and 35)—are partially controlled
within Acp’s, as these genes share a common tissue type (male
accessory glands), similar (high) expression levels, and similar
Because there is reason to believe that both protein evolution
and optimal codon usage are related to gene length (44, 45) and
recombination rate (refs. 25 and 45, and see above), we tested the
possibility that the correlation between dN and Fop is an artifact
of one of these other relationships. We find that optimal codon
usage is significantly correlated with gene length (D. melano-gaster: rs ϭ Ϫ0.142, P ϭ 0.0287; D. simulans: rs ϭ Ϫ0.129, P ϭ
0.0463), as seen in previous studies (42, 43), but not with
recombination (D. melanogaster: rs ϭ Ϫ0.066, P Ͼ 0.05; D.simulans: rs ϭ 0.081, P Ͼ 0.05). [The previously reported
correlation between optimal codon usage and recombination is
weak and detected in a much larger data set than ours (26, 45).]
To distinguish the effects of gene length and protein evolution
on optimal codon usage, we estimated partial correlation coef-
ficients. Both relationships persist, but the correlation between
dN and Fop is much stronger (partial r for gene length vs. dN in
Fop, the frequency of optimal codon usage, vs. dN in D. melanogasterD. melanogaster: Ϫ0.163 vs. Ϫ0.505; in D. simulans: Ϫ0.152 vs.
(a) and D. simulans (b).
Ϫ0.518; gene length and dN are log-transformed; P Ͻ 0.05
substitutions reduces the rate of substitution of linked weakly
Several workers (19, 46, 47) have observed high nonoptimal
favored mutations. This effect is caused by genetic hitchhiking;
codon usage in rapidly evolving genes, but suggested relaxation
as the frequency of hitchhiking increases, weakly selected linked
of selective constraints as the cause. These relaxed constraints
sites behave more neutrally and thus come to more closely reflect
explanations come in two flavors. The first invokes codon-
the mutational spectrum. Because there are more ways, on
specific constraints. Akashi (48) has argued that selection for
average, to mutate to nonoptimal codons, the net effect
translational accuracy (i.e., selection against misincorporation of
of hitchhiking is to increase the fixation rate of nonoptimal
amino acids) should be strongest at functionally important
residues. Consistent with this, he found that evolutionarily
conserved residues tend to use preferred codons, which are less
We test this prediction by asking whether the frequency of
often mistranslated (48). This explanation alone cannot account
optimal codon usage (Fop) decays as the rate of protein evolution
for the results reported here, because even conserved sites in our
increases. As Fig. 2 shows, Fop declines sharply as dN increases
rapidly evolving genes show poor optimal codon usage. After
(statistics identical for both species: rs ϭ Ϫ0.559, P Ͻ 0.0001; the
expunging all divergent codons from the genes in our data, we
same patterns appear using dN͞dS ratios, not shown). dS shows
find that the relationship between F
a weak positive correlation with Fop and dN is unchanged (D.op (D. melanogaster: rs ϭ 0.172,
P Ͻ 0.0061; D. simulans: rs ϭ Ϫ0.546, P Ͻ 0.0001; D. simulans: rs ϭ Ϫ0.555,
s ϭ 0.224, P ϭ 0.0004), but, as partial
correlation analysis shows, the relationship between Fop and dN
The second relaxed constraints explanation invokes gene-
is independent of dS and twice as strong (D. melanogaster: partial
specific constraints, in which constraints on amino acid sites and
r ϭ Ϫ0.505 vs. 0.251; D. simulans: partial r ϭ Ϫ0.491 vs. 0.180).
synonymous sites are correlated within a gene, i.e., genes evolv-
The effect of strongly selected traffic on linked synonymous sites
ing rapidly because of relaxed selection on amino acid compo-
can be illustrated in another way. In Drosophila, AT-biased
sition are likely to also have relaxed selection for optimal codon
mutation pressure causes GC-content at mutational equilibrium
usage (49). There are three reasons to think this explanation
to approach Ϸ35% (42, 43). Coding sequences are nevertheless
does not explain our results. First, the rapid evolution in our data
highly GC-biased, at least partly because of constraints on codon
set does not appear to be caused by relaxed purifying selection,
usage as virtually all preferred codons end in C or G. If a stream
as evidenced by the relationship between rates of recombination
of strongly selected amino acid traffic depresses Ne at weakly
and protein evolution (barring some spurious relationship be-
selected linked synonymous sites, GC-content at those sites
tween high recombination rate and relaxed constraint). Second,
should more closely reflect the mutational spectrum. We find
if the rapid evolution in our data were mostly caused by relaxed
13618 ͉ www.pnas.org͞cgi͞doi͞10.1073͞pnas.212277199
constraints, GC content in both synonymous and amino acid sites
high recombination. There is some indication, though, that
should decay in those loci with high dN͞dS because of AT-biased
genomes are not luxuriant. A comparative quantitative trait
mutation pressure. Although dN͞dS is negatively related to GC
locus study in domesticated cereal species found that convergent
content of synonymous sites (see above), no such negative
traits map to homeologous genomic regions (and therefore
relationship exists for amino acid sites (D. melanogaster: rs ϭ
possibly to the same genes), suggesting that there may be a
0.204, P ϭ 0.0013; D. simulans: rs ϭ 0.214, P ϭ 0.0007). Third,
limited number of ways to construct these traits genetically (51).
interference is the only explanation for why genes with dN͞dS Ͼ
More convincingly, high resolution molecular and experimental
1 (i.e., genes whose rapid evolution is caused by positive selec-
evolution studies have uncovered convergence at the DNA
tion, not relaxed constraints) should show depressed codon
sequence level (52–54), suggesting that selection at least some-
usage: we find that mean Fop for loci with dN͞dS Ͼ 1 is
times uses the same nucleotide repeatedly. If the Drosophila
significantly lower than that for loci with dN͞dS Ͻ 1 (D. mela-
genome is not luxuriant, then our results imply that flies are not
nogaster: Fop, dN/dS Ͻ 1 ϭ 0.534 Ϯ 0.009 vs. Fop, dN/dS Ͼ 1 ϭ 0.355 Ϯ
perfectly adapted because of the slower average response of
0.023, t test ϭ 6.645, P Ͻ 0.0001; D. simulans: Fop, dN/dS Ͻ 1 ϭ
genes in regions of low recombination to directional selection.
0.544 Ϯ 0.009 vs. Fop, dN/dS Ͼ 1 ϭ 0.356 Ϯ 0.022, t test ϭ 7.077, P Ͻ
The weakly selected silent sites of rapidly evolving genes, in
0.0001). Even when we consider only those loci with dN͞dS
contrast, seem more clearly maladapted. With perfect recombi-
significantly greater than 1 (a conservative standard), Fop re-
nation, rapidly evolving genes could both substitute beneficial
mains significantly depressed (D. melanogaster: Fop, dN/dS Ͻ 1 ϭ
amino acids and maintain optimal codon usage.
0.516 Ϯ 0.009 vs. Fop, dN/dS Ͼ 1 ϭ 0.411 Ϯ 0.084, t test ϭ 2.999, P ϭ
The detrimental effects of interference appear hierarchical in
0.0020; D. simulans: Fop, dN/dS Ͻ 1 ϭ 0.523 Ϯ 0.009 vs. Fop, dN/dS Ͼ 1
that linkage constrains protein adaptation, which in turn con-
ϭ 0.415 Ϯ 0.077, t test ϭ 2.878, P ϭ 0.0044). Taken together, these
strains codon adaptation. Formally, either clonal or ruby-in-the
lines of evidence suggest that neither flavor of relaxed constraint
rubbish interference can limit protein evolution. But ruby-in-
hypothesis accounts for the correlation between rapid protein
the-rubbish seems likely more important as the rate of delete-
evolution and low optimal codon usage. Instead, it seems that
rious mutation (55–57), and so the opportunity for interference
interference from strongly selected traffic compromises weakly
from deleterious mutations, far exceeds the rate of favorable
selected codon usage at tightly linked sites.
mutation. Furthermore, low recombination regions in Drosoph-ila may suffer an additional load of deleterious mutations
Concluding Remarks. We find evidence that linkage constrains—
because of the higher numbers of transposable element inser-
and recombination facilitates—adaptation in Drosophila. We
tions found there (58). Assuming that adaptive protein diver-
have shown that (i) the rate of protein adaptation appears
gence is mostly limited by linkage to deleterious mutations, the
limited by interference in low recombination regions, and (ii)
hierarchical effects of interference may reflect the relative
strong directional selection on proteins interferes with weak
magnitudes of selection coefficients in Drosophila: mean selec-
selection for optimal codon usage at linked sites. That such
limits on adaptation are detectable even in a recombining
tion against deleterious mutations is probably stronger than that
genome is surprising and has several implications. It follows,
favoring beneficial amino acid mutations, which in turn is larger
for example, that the limiting effects of linkage on protein
than that favoring preferred codons (i.e., sd Ϸ 10Ϫ2 Ͼ sb,amino acid
adaptation may be manifest in the genetic basis of phenotypic
Ϸ 10Ϫ3 to 10Ϫ4 Ͼ sb,codon Ϸ 10Ϫ6; see refs. 34, 56, and 59). This
evolution. As Birky and Walsh (50) point out in their classic
does not, however, mean that nonoptimal codon usage has a
study of the theory of linkage and selection, ‘‘recombination
negligible effect on genomes. Although selection on individual
enhances the rate of phenotypic evolution, to the extent that
preferred codons is weak (33), the cumulative effect of many
phenotypic evolution is driven by the fixation of advantageous
unpreferred codons may be considerable (33, 37).
mutations.’’ We might expect, therefore, that adaptive species
differences should map disproportionately to high recombi-
We thank P. Andolfatto, D. Begun, J. Bollback, Y. Chen, J. Gillespie, J.
Huelsenbeck, C. Jones, Y. Kim, W. Stephan, two anonymous reviewers,
and especially A. Orr for helpful comments and discussion. This work was
It does not necessarily follow, however, that genes in low
supported by National Institutes of Health Grant GM526738 and by
recombination regions are maladapted. If genomes are luxuriant,
funding from the David and Lucile Packard Foundation (to A. Orr), and
so that there are many ways to adapt to new environments,
by Caspari Fellowships and Messersmith Fellowships (to A.J.B. and
adaptation will simply proceed via substitutions in regions of
1. Fisher, R. A. (1930) The Genetical Theory of Natural Selection (Oxford Univ.
19. Begun, D. J., Whitley, P., Todd, B. L., Waldrip-Dail, H. M. & Clark, A. G.
(2000) Genetics 156, 1879–1888. EVOLUTION
2. Manning, J. T. & Thompson, D. J. (1984) Acta Biotheor. 33, 219–225.
20. Swanson, W. J., Clark, A. G., Waldrip-Dail, H. M., Wolfner, M. F. & Aquadro,
3. Peck, J. (1994) Genetics 137, 597–606.
C. F. (2001) Proc. Natl. Acad. Sci. USA 98, 7375–7379.
4. Orr, H. A. (2000) Genetics 155, 961–968.
21. Wolfner, M. F. (1997) Heredity 88, 85–93.
5. Muller, H. J. (1932) Am. Nat. 66, 118–138.
22. Yang, Z. (1997) Comput. Appl. Biosci. 13, 555–556.
6. Crow, J. F. & Kimura, M. (1965) Am. Nat. 99, 439–450.
23. Goldman, N. & Yang, Z. (1994) Mol. Biol. Evol. 11, 725–736.
7. Gerrish, P. J. & Lenski, R. E. (1998) Genetica 102͞103, 127–144.
24. Ikemura, T. (1981) J. Mol. Biol. 151, 389–409.
8. de Visser, J. A. G. M., Zeyl, C. W., Gerrish, P. J., Blanchard, J. L. & Lenski,
25. McVean, G. A. T & Charlesworth, B. (1999) Genet. Res. 74, 145–158.
R. L. (1999) Science 283, 404–406.
26. Kliman, R. M. & Hey, J. (1993) Mol. Biol. Evol. 10, 1239–1258.
9. Miralles, R., Gerrish, P. J., Moya, A. & Elena, S. F. (1999) Science 285, 1745–1747.
27. Marais, G., Mouchiroud, D. & Duret, L. (2001) Proc. Natl. Acad. Sci. USA 98,
10. Miralles, R., Moya, A. & Elena, S. F. (2000) J. Virol. 74, 3566–3571.
11. Hill, W. G. & Robertson, A. (1966) Genet. Res. 8, 269–294.
28. True, J. R., Mercer, J. M. & Laurie C. C. (1996) Genetics 142, 507–523.
12. Felsenstein, J. (1974) Genetics 78, 737–756.
29. Hilton, H. R., Kliman, R. M. & Hey, J. (1994) Evolution (Lawrence, Kans.) 48,
13. Lewontin, R. C. (1974) The Genetic Basis of Evolutionary Change (Columbia
30. Pal, C., Papp, B. & Hurst, L. D. (2001) Mol. Biol. Evol. 18, 2323–2326.
14. Begun, D. J. & Aquadro, C. F. (1992) Nature 356, 519–520.
31. Comeron, J. & Kreitman, M. (2000) Genetics 156, 1175–1190.
15. Nachman, M. (1997) Genetics 147, 1303–1316.
32. Bachtrog, D. & Charlesworth, B. (2002) Nature 416, 323–326.
16. Stephan, W. & Langley, C. H. (1998) Genetics 150, 1585–1593.
33. Powell, J. R. & Moriyama, E. N. (1997) Proc. Natl. Acad. Sci. USA 94,
17. Nachman, M. W., Bauer, V. L., Crowell, S. L. & Aquadro, C. F. (1998) Genetics150, 1133–1141.
34. Akashi, H. & Eyre-Walker, A. (1998) Curr. Biol. 8, 688–693.
18. Dvorak, J., Luo, M.-C. & Yang, Z.-L. (1998) Genetics 148, 423–434.
35. Kreitman, M. & Antezana, M. (2000) in Evolutionary Genetics: From Molecules
PNAS ͉ October 15, 2002 ͉ vol. 99 ͉ no. 21 ͉ 13619 to Morphology, eds. Singh, R. S. & Krimbas, C. B. (Cambridge Univ. Press, New
50. Birky, C. W. & Walsh, J. B. (1988) Proc. Natl. Acad. Sci. USA 85, 6414–6418.
51. Paterson, A. H., Yann-Rong, L., Zhikang, L., Schertz, K. F., Doebley, J. F.,
36. Akashi, H. (1995) Genetics 139, 1067–1076.
Pinson, S. R. M., Liu, S.-C., Stansel, J. W. & Irvine, J. E. (1995) Science 269,
37. Barton, N. H. (1995) Genetics 140, 821–841.
38. Stephan, W., Charlesworth, B. & McVean, G. (1999) Genet. Res. 73, 133–146.
52. Stewart, C.-B. & Wilson, A. C. (1987) Cold Spring Harbor Symp. Quant. Biol.
39. McVean, G. A. T. & Charlesworth, B. (2000) Genetics 155, 929–944. 52, 891–899.
40. Barton, N. H. (1994) Genet. Res. 64, 199–208.
53. ffrench-Constant, R. H. (1994) Insect Biochem. Mol. Biol. 24, 335–345.
41. Gillespie, J. H. (2001) Evolution (Lawrence, Kans.) 55, 2161–2169.
54. Wichman, H. A., Badgett, M. R., Scott, L. A., Boulianne, C. M. & Bull, J. J.
42. Moriyama, E. N. & Hartl, D. L. (1993) Genetics 134, 847–858.
(1999) Science 285, 422–424.
43. Petrov, D. A. & Hartl, D. L. (1999) Proc. Natl. Acad. Sci. USA 96, 1475–1479.
55. Mukai, T., Chigusa, S. I., Mettler, L. E. & Crow, J. F. (1972) Genetics 72,
44. Marais, G. & Duret, L. (2001) J. Mol. Evol. 52, 275–280.
45. Comeron, J. M., Kreitman, M. & Aguade, M. (1999) Genetics 151, 239–249.
56. Keightley, P. D. & Eyre-Walker, A. (1999) Genetics 153, 515–523.
46. Schmidt, P. S., Duvernell, D. D. & Eanes, W. F. (2000) Proc. Natl. Acad. Sci.
57. Lynch, M., Blanchard, J., Houle, D., Kibota, T., Schultz, S., Vassilieva, L. &
USA 97, 10861–10865.
Willis, J. (1999) Evolution (Lawrence, Kans.) 53, 645–663.
47. Schmid, K. J. & Aquadro, C. F. (2001) Genetics 159, 589–598.
58. Charlesworth, B. (1996) Genet. Res. 68, 131–150.
48. Akashi, H. (1994) Genetics 136, 927–935.
59. Gillespie, J. H. (1991) The Causes of Molecular Evolution (Oxford Univ. Press,
49. Comeron, J. M. & Kreitman, M. (1998) Genetics 150, 767–775. 13620 ͉ www.pnas.org͞cgi͞doi͞10.1073͞pnas.212277199
ST 13-0068-GIL 11/27/2013 CONSTRUCTION CONTRACTORS Construction contractors who physically incorporate tangible personal property into real estate owned by exem pt organizations or governmental entities that hold tax exempt “E” numbers can purchase such property tax free by providing their suppliers with the certification described in 86 Ill. Adm. Code 130.2075(d). See 86 Ill. Adm. Code Sect
 For the Y and dot-fourth chromosomes, we assume recombina-
tion is absent. The mean recombination rate in this data set is c ϭ
0.0029 centimorgan (cM)͞kb, close to the global mean for the D.
melanogaster genome (see supplimentary information for ref.
For the Y and dot-fourth chromosomes, we assume recombina-
tion is absent. The mean recombination rate in this data set is c ϭ
0.0029 centimorgan (cM)͞kb, close to the global mean for the D.
melanogaster genome (see supplimentary information for ref.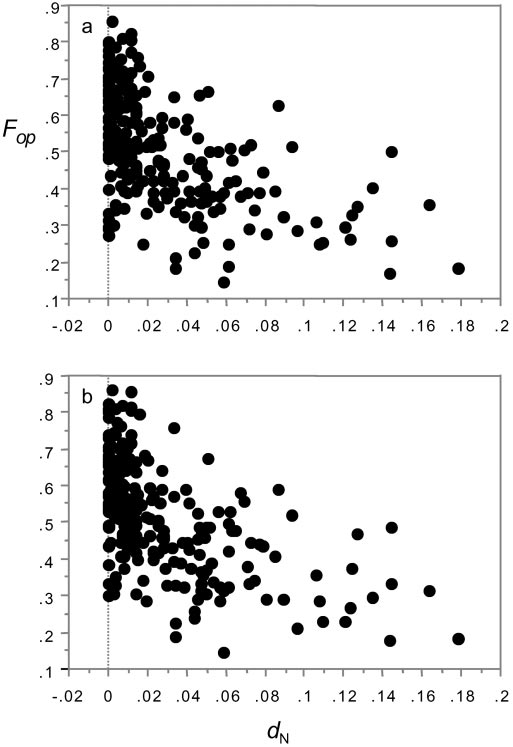 that this is indeed the case. As the rate of amino acid substitution
increases, third position GC-content decreases significantly (dN,
D. melanogaster: rs ϭ Ϫ0.540, P Ͻ 0.0001; dN, D. simulans: rs ϭ
Ϫ0.549, P Ͻ 0.0001; dN͞dS, D. melanogaster: rs ϭ Ϫ0.569, P Ͻ
0.0001; dN͞dS, D. simulans: rs ϭ Ϫ0.584, P Ͻ 0.0001).
that this is indeed the case. As the rate of amino acid substitution
increases, third position GC-content decreases significantly (dN,
D. melanogaster: rs ϭ Ϫ0.540, P Ͻ 0.0001; dN, D. simulans: rs ϭ
Ϫ0.549, P Ͻ 0.0001; dN͞dS, D. melanogaster: rs ϭ Ϫ0.569, P Ͻ
0.0001; dN͞dS, D. simulans: rs ϭ Ϫ0.584, P Ͻ 0.0001).